Splitting Fastq Formatted Files
Most NGS sequencing platforms have the ability to save data in Fastq formatted files. Typically, these are provided as a pair of files, representing paired-end reads, though these may also be provided as interleaved reads in a single file. Depending on the source, these files can have just a few hundred reads up to as many as 50 or 100 million. There are many occasions where the number of reads in these files is not only overkill for the required experiment, but can actually prevent algorithms from correctly assembling them into appropriate contigs. Ideally, for de novo assembly, you want no more than 100-500x coverage of the genome. If you are sequencing a small plasmid, this may mean just a few thousand reads in the data files, even if your sequencing proider has sent data files containing many millions of reads.
In addition, working with smaller numbers of reads reduces computation, memory and disk space requirements - MacVector can often assemble entire bacterial genomes in just a few minutes with an optimized set of reads whereas the full set may take many hours, especially on Macs equipped with less than 16 GB RAM.
We at MacVector have created a simple free utility called SplitFastqFile.app to simplify splitting large files into smaller files. You can download a zipped copy of the utility using this link.
Using SplitFastqFile.app
SplitFastqFile.app is a "droplet" application, meaning that after you download and extract the application, you just need to drag a fastq formatted file onto it and it will split that file into multiple smaller files. During the splitting, you will get prompted for various inputs;
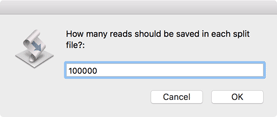
First, you should choose how many reads to save in each split file. Aim for a coverage of between 100x and 500x for the molecule you are sequencing, but be aware that if you have a pair of files, you should use the same value for both files.
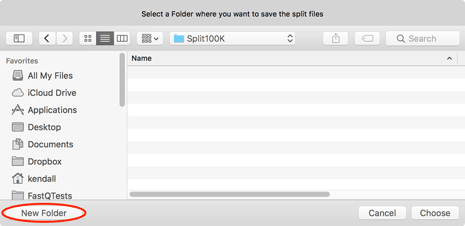
Next you will be prompted for a destination - be sure to use the New Folder button if you want to save the files in a unique location.
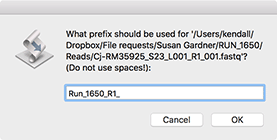
Now select a "Prefix" for the filenames. This will be used as the root of the filename, so if you choose (like here) "Run_1650_R1_" than the files will be named "Run_1650_R1_aaa.fastq", "Run_1650_R1_aab.fastq" and so on.
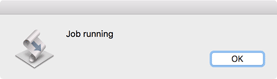
A dialog will appear while the job is running. With long jobs this will close and reopen multiple times. For very large files (>50 million reads) the job may take several minutes to run on a typical machine. Be patient!
Finally, you will get a dialog box confirming that the utility has finished processing the files.
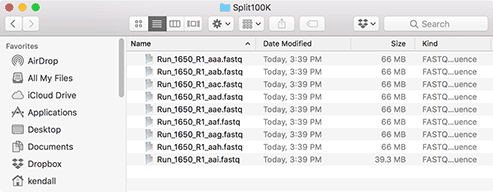
The target folder will contain all of the split files, as shown here. The original file is never changed by the utility.
|
